
In the mid-80s, Syvanen postulated that the exchange of genetic material between organisms (Horizontal Gene Transfer, HGT) is an important and widespread evolutionary mechanism (Syvanen 1985), and summarized the evidence for that conclusively in the mid-90’s (Syvanen 1994). After that, HGT rapidly became a major focus of research, which showed that the process is ubiquitous in all domains of life including eukaryotes, and significantly shifted our understanding of how genomes evolve (Keeling 2024). However, the results to date show a highly non-uniform landscape: while all domains of life seem to have organisms that readily incorporate foreign genetic material, all have organisms that rarely or never do it.
Our article is a part of the current attempts to gain a more overarching insight into the complex landscape of HGT in eukaryotes. On a broader scale, how often do these organisms incorporate foreign genes into their genomes, and what happens with them afterwards – both with the organisms, and with the genes? Does the organism’s ecological niche or taxonomic placement influence whether it readily participates in genetic exchange, or rather prefers genomic stability? How often is HGT a real “evolutionary shortcut”, and how often just a genomic noise?
To pick a representative group of organisms for our research, we focused on the fungi. These organisms are remarkably diverse, from aquatic parasites of animals, through terrestrial decomposers of dead organic matter, to anaerobic symbionts of herbivores. Not to mention their commercial and medical importance: they cause severe diseases with a total death toll greater than malaria. And some of them do participate in genetic exchange in remarkable ways (Gonçalves, C., Hittinger, C.T., Rokas, A. 2024), including giant transposons that massively move genes across diverse filamentous fungi (Bucknell and McDonald 2023).
We focused on 44 fungi belonging to the so-called early diverging group, which represent the most diverse fungal lineages and are severely understudied. To find horizontally acquired genes, we developed a methodology of comparative studies of HGT in eukaryotes and implemented it in a computational pipeline. We have identified several evolutionary scenarios in which certain fungal genomes became enriched in xenologs, with varying rates of participation in genetic exchange and duplication of the received genetic material, resulting in enriched metabolic, adaptive, and immune capabilities. The process is often associated with gene fusion, as the foreign genetic material can get incorporated within a pre-existing gene. Afterward, a foreign protein-coding gene can even obtain fragments of other genes of the host through mechanisms such as the shuffling of protein domains, which can further modify its functions, a result that combines, unifies, and extends previous observations (Culbertson and Levin 2023).
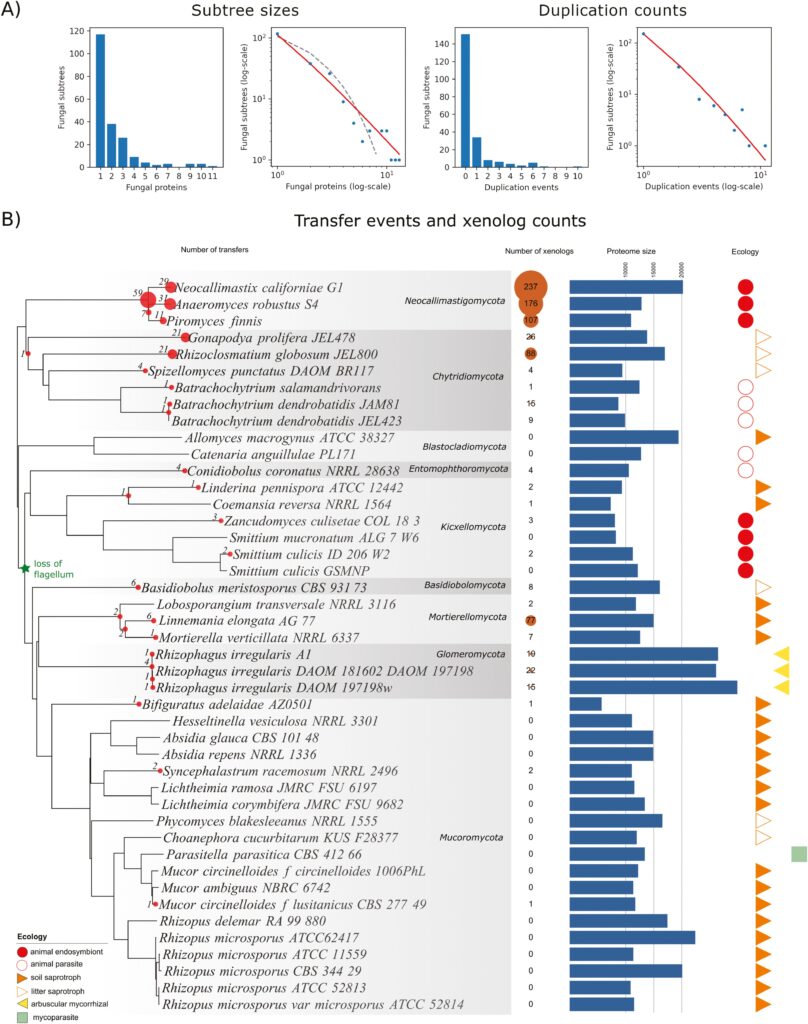
Our results paint a detailed, intricate landscape of genetic exchange throughout the fungal tree of life. However, it wasn’t easy to arrive at this point. The project started in 2014, and what was seemingly a simple idea turned into an almost ten-year-long journey of repeatedly checking the results, making necessary improvements to the pipeline, re-calculating the results, and finding even more problems. As we show in our article, there is a particular, multi-layered complexity to eukaryotic HGT, one that is on the verge of the capabilities of current algorithms, necessitating a tailored approach that overcomes numerous pitfalls and limitations.
Our initial approach seemed reasonable: use compositional statistics such as an atypical GC content and Codon Adaptation Index to get an initial set of potentially foreign, protein-coding genes; use BLAST to detect homologs of the encoded proteins; cluster and align the sequences; infer maximum likelihood phylogenetic trees; and finally, use the tree reconciliation algorithms to compare the gene trees to the species trees from NCBI Taxonomy. We implemented this approach, et voilà, we obtained a set of poorly supported trees littered with chains of implausible transfers across kingdoms.
Which step was the culprit? Every single one. Starting with tree reconciliation algorithms, which look for HGT events by embedding a gene tree into the corresponding species tree. Unbeatable in analyses of individual genes, it turned out they struggle with large-scale comparative studies. First, they require the user to specify the “evolutionary costs” of gene transfers, arbitrary parameters that do not have a clear biological interpretation nor established methods to estimate them in a high throughput setting. Second, as our results eventually confirmed, we suspected that transfer rates may differ markedly between organisms, violating the implicit assumption of a constant “cost” of transfer. These and other problems prompted us to develop an algorithm that can detect incongruencies between trees without the need to specify cost parameters (Ciach, Muszewska, and Górecki 2018).
It then turned out that neither reconciliation nor our approach was sufficient for the task at hand. Both approaches had a particular requirement that we couldn’t satisfy: accurately rooted gene trees. In a high throughput setting, outgroups are unavailable, and the accuracy of outgroup-free rooting methods turned out to be gravely insufficient for our needs. Thus, we needed to develop yet another approach, one that can work with unrooted trees. We based our idea on the way trees are often analyzed manually: check which species are on each side of a fungal subtree and compare them to the species tree. This finally led us to an algorithm that worked for our study.
One that worked on correct input data, that is. We then discovered problems much deeper in the pipeline: the clustering software struggled. As we show in our article, protein sequences can lose or gain up to 400 amino acids during or after transfer and are often fused with fungal genes, leading to an inconsistent homology and consequently a quasi-random assignment to clusters.
After eliminating the problems one by one, we arrived at the very bottom of our pipeline: filters based on compositional statistics discarded many notable cases of HGT while selecting many vertically inherited genes as “potentially interesting”. In hindsight, this is not surprising: as we show in our article, many bacterial genes quickly ameliorate into the host genomes, leading to, for example, bacteria-derived genes with up to 14 introns. Ancestrally aquatic fungi turned out to be generally more likely to acquire foreign genetic material than terrestrial ones. Endosymbiotic bacteria were revealed to be a source of useful xenologs, as exemplified by NOD-like receptors transferred to Mortierellomycota.

Finally, we arrived at our destination. While there is still much to be done, we finally get a glimpse at the complex landscape of HGT across diverse fungal species, the intricate complexity of its mechanisms, and the diverse ways it can result in evolutionary advantages. We describe more details, including the taxonomic, genomic, and molecular aspects of fungal HGT, in our article.
References:
Ciach, Michał Aleksander, Anna Muszewska, and Paweł Górecki. 2018. “Locus-Aware Decomposition of Gene Trees with Respect to Polytomous Species Trees.” Algorithms for Molecular Biology: AMB 13 (June): 11.
Culbertson, Edward M., and Tera C. Levin. 2023. “Eukaryotic CD-NTase, STING, and Viperin Proteins Evolved via Domain Shuffling, Horizontal Transfer, and Ancient Inheritance from Prokaryotes.” PLoS Biology 21 (12): e3002436.
Gonçalves, C., Hittinger, C.T., Rokas, A. 2024. “Horizontal Gene Transfer in Fungi and Its Ecological Importance.” In Fungal Associations, edited by Hsueh Y P Blackwell. Vol. 9. The Mycota. Springer.

