A new Comment & Opinion piece published in Evolution Letters provides a roadmap for analysing genetic data in the context of sex-specific adaptation. Here, lead author Filip Ruzicka explains the motivation for and key conclusions of the study.
Sexual dimorphism—or differences in female and male traits—represents the most striking instance of adaptive differentiation within single species. Familiar examples of sexual dimorphism include the ornate tails of peacocks, the bright plumage of male birds of paradise, and the exaggerated eye stalks of stalk-eyed flies. The ultimate cause of sexual dimorphism, as initially put forward by Darwin*, is sex-specific selection**—a broad term that encompasses sexually antagonistic selection (in which the sexes are subject to opposing directions of selection), sex-limited selection (selection occurs in one sex only) and sexually concordant selection (the direction of selection is aligned between the sexes, though the strength of selection may differ between them).
Sex-specific selection fascinates evolutionary biologists for a number of reasons. In addition to its relevance to sexual dimorphism, it provides clues to the maintenance of genetic variation for fitness, aspects of genome evolution, and even population viability. For example, genetic variants with opposing fitness effects in each sex (“sexually antagonistic alleles”) can persist much longer than alleles that are deleterious or beneficial for both sexes, and may account for the puzzlingly high levels of standing genetic variation for fitness that populations often harbour. The localisation of sex-specific variants on sex chromosomes vs. autosomes has also been a source of much research interest, and may initiate sex chromosome degeneration, as observed on Y chromosomes (in mammals and fruit flies) and W chromosomes (in butterflies and birds). Sex-specific selection can even aid species persistence in cases where genetic variation is overwhelmingly sexually concordant and selection is stronger on males than females.
Despite strong interest among evolutionary biologists, we still know remarkably little about the genetic basis of sex-specific adaptation. To some extent, this is understandable. The application of genomics to adaptation, including sex-specific adaptation, is recent and primarily spans the last decade or so. Genomic approaches must also overcome the considerable challenge of separating signal from noise (i.e., sampling error) and controlling for processes that superficially resemble selection (e.g., uncontrolled population structure and technical artefacts). Furthermore, studying the genomics of sex-specific adaptation presents some unique difficulties. For example, exceptionally pronounced allele frequency differences between individuals sampled from different geographic localities within a species’ range can indicate local adaptation, whereas sexual reproduction constrains males and females to be part of the same population, thereby limiting opportunities for allele frequency differentiation between the sexes.
Two factors motivated us to undertake the new study published in Evolution Letters. First, we were keen to clarify the potential for genomic approaches to detect signals of sex-specific selection. Second, our colleagues in the related fields of local adaptation and statistical genomics have spent decades developing methods to more accurately identify the genetic basis of local adaptation and quantitative trait variation. It therefore seemed timely to draw insights from these more mature fields and apply them in genomic studies of sex-specific adaptation.
In our study, we distinguish between two basic approaches to identify genes and genome-wide signals associated with sex-specific adaptation: (i) indirect approaches, which primarily use allele frequency differences between the sexes (FST and related metrics of allele frequency differentiation), and (ii) direct approaches, which use associations between alleles and fitness measurements.
Two important conclusions can be drawn from our study (but see the publication for more details). First, models show that opportunities for identifying individual candidate loci under sex-specific selection using allele frequency differentiation between the sexes are severely limited, except for very large datasets (realistically, sample sizes of 104 or larger). Nevertheless, re-analyses reveal that polygenic signals of sex-specific selection are detectable in 2 of 3 previously published datasets (i.e., allele frequency differentiation between the sexes cannot be accounted for by random sampling effects alone), even in the absence of individually significant “hits” (Figure 1). Though population structure or other technical artefacts may contribute to the inflated allele frequency differences between sexes in these datasets, we highlight approaches that minimise this eventuality (e.g., case-control GWAS, stringent quality-filtering, functional validation of candidate loci).
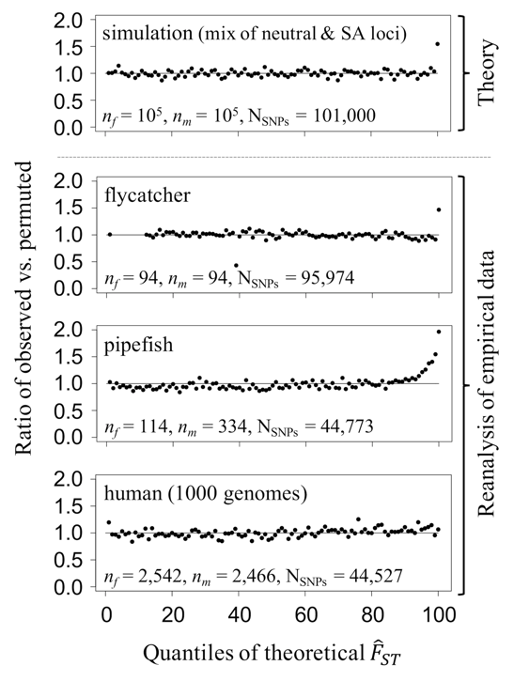
Figure 1. Polygenic signals of sex-specific selection in genomic data. In each quantile of the null distribution of FST (x axis), we calculated the proportion of loci (top panel, simulated; bottom panels, empirical data) that fall into a given quantile of the null distribution of FST (y axis) before and after permutation of sex labels, thus obtaining a ratio of observed to permuted proportions. In pipefish and flycatcher datasets, an excess of observed loci in the tails of the theoretical null distribution of FST (right), suggesting signals of sex-specific selection. Figure is from Ruzicka et al. 2020 (Evolution Letters).
Second, empirical approaches that identify candidate loci underlying sex-specific adaptation by testing for direct associations between alleles and measurements of sex-specific fitness are severely under-utilised. We review the state of the field on this front, including candidate loci that have (to date) been identified using direct approaches, and highlight a range of empirical systems and approaches that have particularly strong potential for future research on the genetics of sex-specific adaptation (Figure 2).

Figure 2. Some example systems amenable to direct approaches for uncovering sex-specific selection (Florida scrub-jays, humans, white campion, common fruit fly, cowpea seed beetle).
These systems include pedigreed populations (where fitness can be measured accurately), Drosophila hemiclone lines (because fitness can be measured across replicate outbred individuals), large biobanks (due to their high power to detect causal loci), experimental systems amenable to artificial selection and re-sequencing (multigenerational responses to sex-specific selection increase statistical power), and certain plant systems (because they are experimentally tractable for field study and/or exhibit heightened population genetic signals of selection by way of enhanced expression or prolonged life-cycles in haploid phase).
We hope our study provides a framework for more research into the genomic basis of sex-specific adaptation.
Filip Ruzicka is a Research Fellow in the School of Biological Sciences, Monash University. Coauthors Tim Connallon and Ludovic Dutoit also contributed to this blog. The original article is freely available to read and download from Evolution Letters.
The study was a collaboration among researchers based in France, Sweden, Switzerland, the UK, Australia and New Zealand. The authors are grateful to the European Society for Evolutionary Biology for funding the “Special Topics Network” workshop which motivated this work.
Footnotes
*Darwin used the term “sexual selection”, which specifically refers to differential selection on components of fitness related to mating success. The term sex-specific selection refers to selection on any component of fitness (e.g. differences in viability between the sexes) and is thus somewhat more general.
**In some contexts, sex-specific selection can refer to sex-limited selection. We instead use it as a synonym for “sex differences in selection”.